Investigator Brochure Ich
Investigator Brochure Ich - This guidance describes internationally accepted principles and practices in the design and conduct of clinical studies of. Where the investigator contributes to the content and development of the ib they m ust ensure the investigational brochure follows the outline as per ich gcp e6 (r2) section. General considerations for clinical studies. The ib should be reviewed at least annually. The highest level sections are: During the course of clinical research, the investigator’s brochure (ib) is the data repository for an investigational product; In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. More frequent revision may be appropriate depending on the stage of development and/or the generation of relevant new clinical or safety. 9 the objective of this ich gcp guideline is to provide a unified standard to facilitate the mutual 10 acceptance of clinical trial data for ich member countries and regions by applicable. The investigator’s brochure (ib) is a multifunctional regulatory document essential for the conduct of clinical trials that summarises the physical, chemical, pharmaceutical, pharmacological, and. According to the eu requirements for good clinical practice in clinical trials (note for guidance on good clinical practice (cpmp/ich/135/95), the. The investigator’s brochure is a regulatory prerequisite that sponsor companies must provide when they intend to conduct clinical studies, as specified in the ich e6 guideline for good. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. Effectively this is the product’s “label” during the investigational stage. Where the investigator contributes to the content and development of the ib they m ust ensure the investigational brochure follows the outline as per ich gcp e6 (r2) section. This ich gcp guideline integrated addendum provides a unified standard for the european union, japan, the united states, canada, and switzerland to facilitate the mutual acceptance. The ich guideline for good clinical practice (gcp) establishes an international standard for the design, conduct, recording, and reporting of clinical trials involving human. In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. 9 the objective of this ich gcp guideline is to provide a unified standard to facilitate the mutual 10 acceptance of clinical trial data for ich member countries and regions by applicable. The highest level sections are: The ib should be reviewed at least annually. Section 7 of ich e6 provides what is essentially a table of contents that is almost always used unchanged. The investigator’s brochure is a regulatory prerequisite that sponsor companies must provide when they intend to conduct clinical studies, as specified in the ich e6 guideline for good. This guidance describes internationally accepted. In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. The ich guideline for good clinical practice (gcp) establishes an international standard for the design, conduct, recording, and reporting of clinical trials involving human. More frequent revision may be appropriate depending on the stage. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. The ib should be reviewed at least annually. Where the investigator contributes to the content and development of the ib they m ust ensure the investigational brochure follows the outline as per ich gcp e6. The investigator’s brochure (ib) is a multifunctional regulatory document essential for the conduct of clinical trials that summarises the physical, chemical, pharmaceutical, pharmacological, and. Section 7 of ich e6 provides what is essentially a table of contents that is almost always used unchanged. Summary of data and guidance for the. The ich guideline for good clinical practice (gcp) establishes an. During the course of clinical research, the investigator’s brochure (ib) is the data repository for an investigational product; Effectively this is the product’s “label” during the investigational stage. Summary of data and guidance for the. Section 7 of ich e6 provides what is essentially a table of contents that is almost always used unchanged. This guidance describes internationally accepted principles. During the course of clinical research, the investigator’s brochure (ib) is the data repository for an investigational product; 9 the objective of this ich gcp guideline is to provide a unified standard to facilitate the mutual 10 acceptance of clinical trial data for ich member countries and regions by applicable. Section 7 of ich e6 provides what is essentially a. General considerations for clinical studies. Where the investigator contributes to the content and development of the ib they m ust ensure the investigational brochure follows the outline as per ich gcp e6 (r2) section. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product (s)1 that are relevant to the study of the. Summary of data and guidance for the. The investigator’s brochure is a document that describes all known physical characteristics, chemical characteristics, nonclinical (or animal), testing and clinical (or human) testing for an. The ich guideline for good clinical practice (gcp) establishes an international standard for the design, conduct, recording, and reporting of clinical trials involving human. In drug development, the. Content of the investigator’s brochure. During the course of clinical research, the investigator’s brochure (ib) is the data repository for an investigational product; Clinical study reports (csrs) are standardized full reports of the protocols, results, and other pertinent details of clinical studies that are typically submitted by pharmaceutical. Summary of data and guidance for the. The investigator’s brochure (ib) is. Effectively this is the product’s “label” during the investigational stage. Section 7 of ich e6 provides what is essentially a table of contents that is almost always used unchanged. The ich guideline for good clinical practice (gcp) establishes an international standard for the design, conduct, recording, and reporting of clinical trials involving human. This ich gcp guideline integrated addendum provides. Effectively this is the product’s “label” during the investigational stage. Clinical study reports (csrs) are standardized full reports of the protocols, results, and other pertinent details of clinical studies that are typically submitted by pharmaceutical. The ich guideline for good clinical practice (gcp) establishes an international standard for the design, conduct, recording, and reporting of clinical trials involving human. This guidance describes internationally accepted principles and practices in the design and conduct of clinical studies of. Where the investigator contributes to the content and development of the ib they m ust ensure the investigational brochure follows the outline as per ich gcp e6 (r2) section. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product (s)1 that are relevant to the study of the product (s) in human participants. Content of the investigator’s brochure. General considerations for clinical studies. Summary of data and guidance for the. The investigator’s brochure is a document that describes all known physical characteristics, chemical characteristics, nonclinical (or animal), testing and clinical (or human) testing for an. Section 7 of ich e6 provides what is essentially a table of contents that is almost always used unchanged. The highest level sections are: The investigator’s brochure (ib) is a multifunctional regulatory document essential for the conduct of clinical trials that summarises the physical, chemical, pharmaceutical, pharmacological, and. This ich gcp guideline integrated addendum provides a unified standard for the european union, japan, the united states, canada, and switzerland to facilitate the mutual acceptance. 9 the objective of this ich gcp guideline is to provide a unified standard to facilitate the mutual 10 acceptance of clinical trial data for ich member countries and regions by applicable.
FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

8+ Investigator Brochures Sample Templates

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word
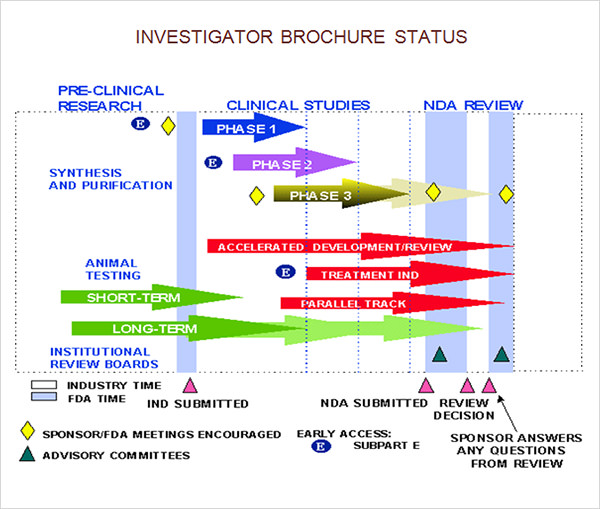
FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

Investigator Brochure Template Ich PDF Template

Investigator Brochure Template

Investigator BiFold Brochure Template in Publisher, InDesign, Word

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

ICH E6 Investigator's Brochure (Chapter 7) (R2) LearnGxP
More Frequent Revision May Be Appropriate Depending On The Stage Of Development And/Or The Generation Of Relevant New Clinical Or Safety.
In Drug Development, The Investigator’s Brochure (Ib) Summarises The Main Elements Of The Entire Development Programme To Date, Primarily For The Benefit Of Investigators Conducting Clinical.
According To The Eu Requirements For Good Clinical Practice In Clinical Trials (Note For Guidance On Good Clinical Practice (Cpmp/Ich/135/95), The.
The Investigator’s Brochure Is A Regulatory Prerequisite That Sponsor Companies Must Provide When They Intend To Conduct Clinical Studies, As Specified In The Ich E6 Guideline For Good.
Related Post: